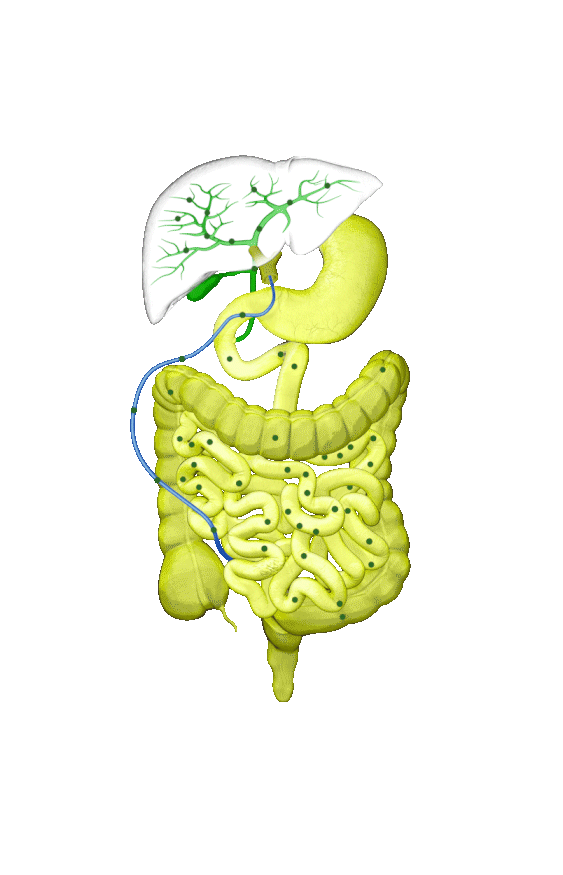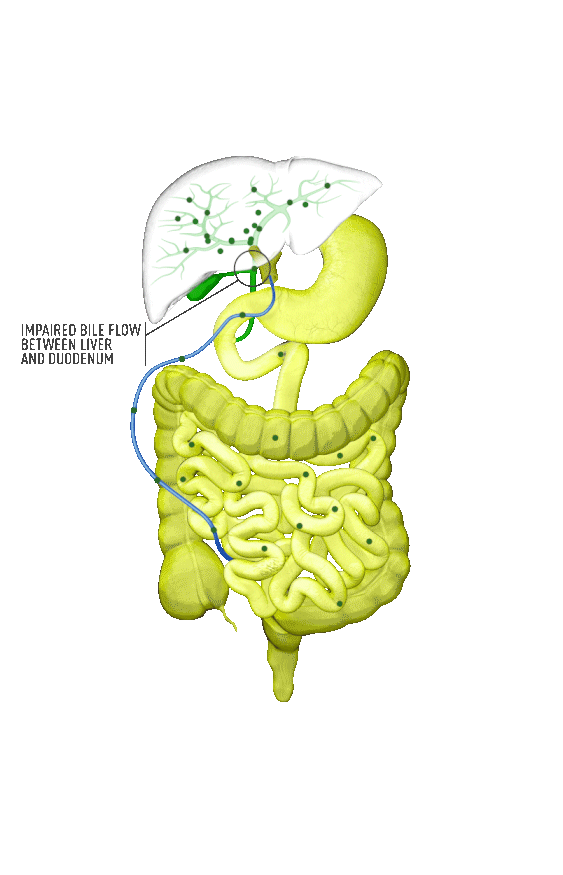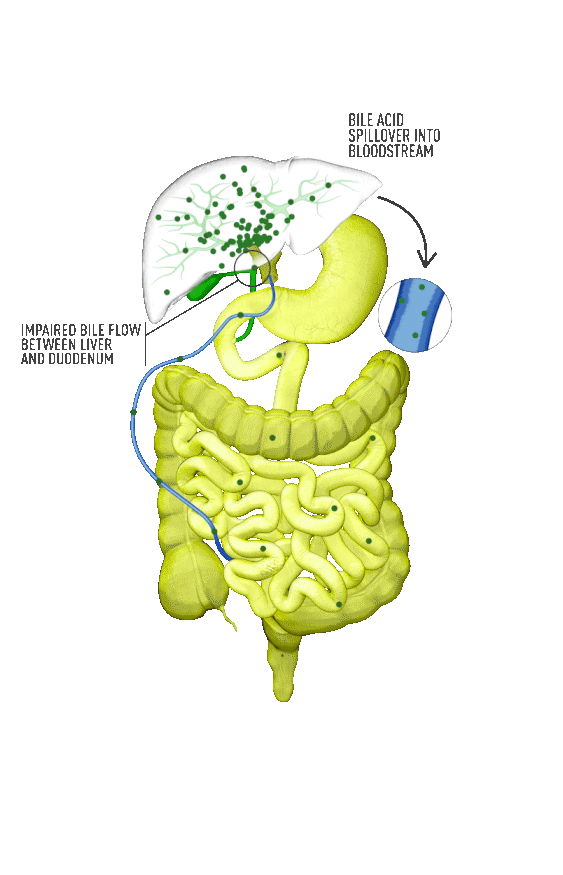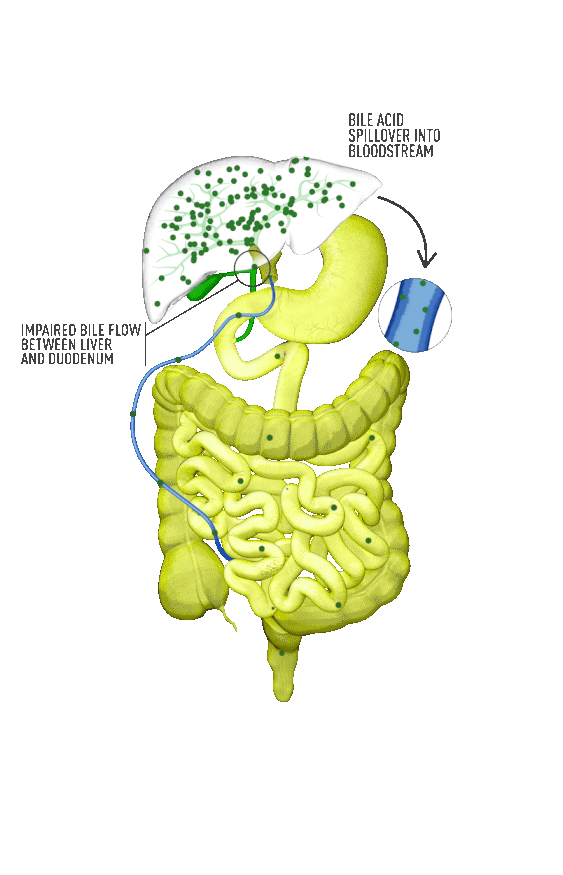
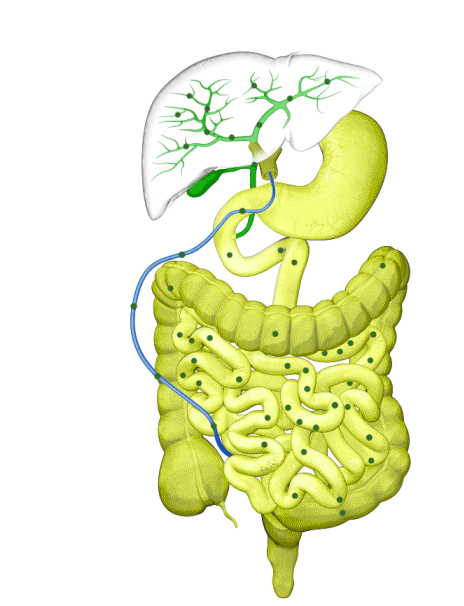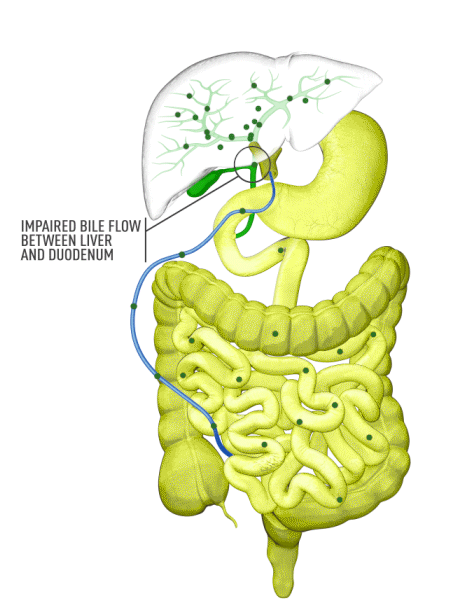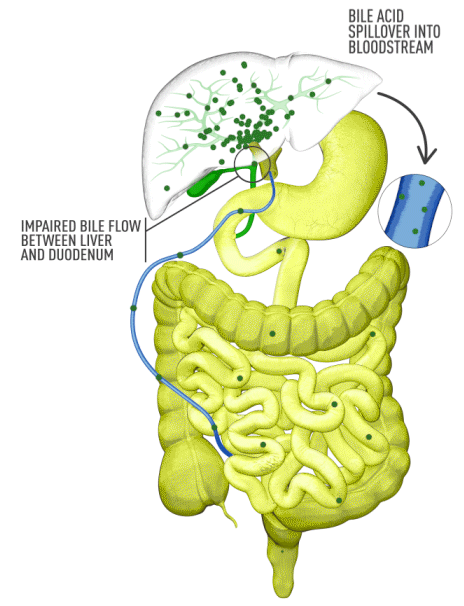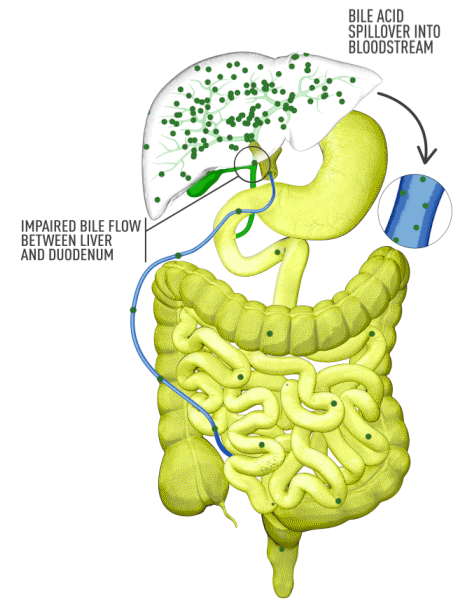
Although the symptoms of cholestatic liver disease may vary based on the etiology, many patients with cholestasis present with intense itchiness, jaundice, dark urine, and pale stools. The impairment of bile flow into the intestine also means that calcium, lipids, and fat-soluble vitamins are poorly absorbed. If cholestasis persists, a deficiency of these nutrients can cause loss of bone tissue.3,4

Chronic cholestatic liver diseases are a significant cause of morbidity and mortality within the pediatric population. The primary cholestatic diseases of infancy and childhood are frequently symptomatic and often rapidly progressive. These chronic cholestatic diseases that present in childhood include biliary atresia, Alagille syndrome, progressive familial intrahepatic cholestasis (PFIC), bile acid synthesis defects, cystic fibrosis-related liver disease, ductal plate abnormalities, primary sclerosing cholangitis (PSC), and certain metabolic diseases.5 This website focuses on Alagille syndrome and PFIC, specifically.

There are several genetic disorders that cause cholestasis, including Alagille syndrome and PFIC. Genetic testing can confirm if a patient is carrying the respective gene and aid in diagnosing patients with these cholestatic syndromes.6 However, there are various investigations that aid in diagnosing cholestatic liver disease.
In Alagille syndrome, variability of clinical presentation and disease severity can result in underdiagnosis, misdiagnosis, or delayed diagnosis. Molecular genetic testing, therefore, provides valuable confirmation, particularly in milder cases or in diagnosis of extended family.7,8
With PFIC, there are several investigations that help to identify its classification into types 1, 2, or 3, and differentiate it from other causes of cholestasis.9 However, no phenotypic features can distinguish PFIC1 from PFIC2. As a result, genetic testing is the only reliable way to distinguish among PFIC subtypes, making it the gold standard in diagnostic tools.9-11
Mirum is proud to sponsor genetic testing through Travere Therapeutics to help with early diagnosis of Alagille syndrome.
The field of inherited cholestatic liver diseases is evolving rapidly as a result of advances in diagnosis based on genetic mutation associations.12